为了进一步学习顶刊文献的研究思想和方法,研之成理推出“顶刊精读”专栏。我们希望能够深入理解这些高质量的论文,把其中蕴含的研究方法/技巧,实验设计理念和构思方法等展示给大家。如果大家碰到不错的文献,欢迎来研之成理投稿,向大家分享自己的收获。今天为大家带来的是发表在Nature上的一篇文章。

▲DOI: 10.1038/s41586-019-1782-2
如何将已发表工作升华至Nature?——分子添加剂调节铜催化CO2电还原产乙烯
发现问题
Jonas C. Peters 和 Theodor Agapi等人(2017)发现在电解质溶液中加入一定量的N-tolylpyridinium盐可以大幅度提高铜表面CO2电还原产乙烯和乙醇的选择性(由21%提升至70%),进一步的研究表明这一性能的提升与N-arylpyridinium盐在铜表面原位形成的二聚体沉淀有关(如图 1)。尽管他们曾试图解释深层原因,但并未发现可信的直接证据。[1]因此,该工作留下的一个未解问题是:为什么这种二聚体沉淀可以提升C2+产物选择性?或者什么因素决定这种沉淀与CO2电还原的关系?解决此问题可以帮助开发更高效的类似N-arylpyridinium盐的添加剂来提高CO2电还原产C2+产物的选择性。
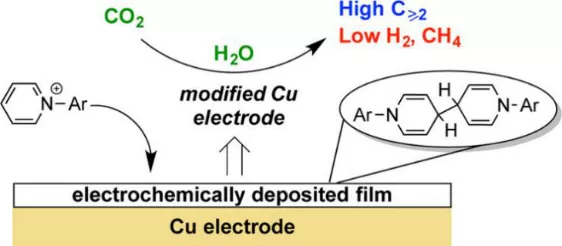
▲图1 多晶铜表面N-tolylpyridinium chloride衍生的二聚体沉淀促进CO2电还原C2+产物的示意图
升华
Edward H. Sargent,Jonas C. Peters和Theodor Agapi等人的这篇Nature文章(2019)的工作核心是找出该问题的答案,即确定关键因素或可操作的描述因子(descriptor),并基于对其中关键因素的理解开发出更有效的电解质添加剂来提高CO2电还原性能。[2]
猜想
前期理论和实验经验表明催化剂表面的局部环境与反应物和中间产物的相互作用可以调节CO2电还原性能,作者通过实验发现N-arylpyridinium盐衍生的二聚体沉淀不影响铜催化剂的表界面性质,例如形貌、晶体结构、电子结构、氧化态、配位环境和浸润性等,也不影响反应过程中所涉及到的诸如反应物、相关离子、氢原子、产物等的传递。因此作者推测N-arylpyridinium盐衍生的二聚体可能通过其与反应中所产生的中间体的相互作用来调节CO2电还原的选择性。
寻找证据
作者通过确定乙烯的选择性(A)、N-arylpyridinium盐衍生的二聚体的给电子能力(B)和电极表面CO的吸附构型比(COatop/CObridge)(C)三者之间相互关系,得出二聚体的给电子能力为决定其对CO2电还原促进的描述因子。
A与B相关:作者合成了11种基于N-arylpyridinium的盐,并计算了它们的给电子能力(由氮原子的Bader charge来衡量),测试了它们对CO2电还原产乙烯的选择性的影响,发现了如图 2所示的乙烯选择性与供电子能力的火山型关系。
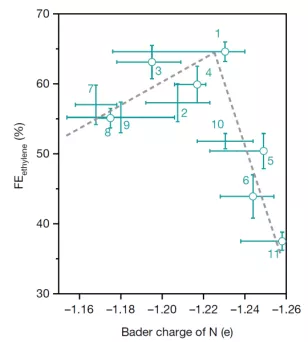
▲图2 乙烯选择性(法拉第效率)随N-arylpyridinium衍生的二聚体中氮原子的给电子能力(Bader charge)的变化趋势
A与C相关:作者用原位拉曼光谱测试了这11种N-arylpyridinium盐对CO在电极表面的吸附构型比(COatop/CObridge)的影响,并得到了乙烯选择性与CO吸附构型比的火山型关系,如图 3。
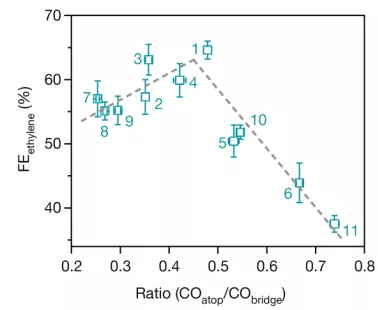
▲图3 N-arylpyridinium衍生的二聚体沉淀在铜表面时,乙烯选择性与CO在电极表面的吸附构型比(COatop/CObridge)的关系。
B与C相关:作者进一步分析发现了CO吸附构型与供电子能力呈线性正相关,如图 4。
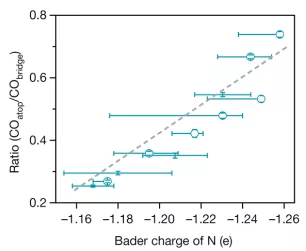
▲图4 CO在电极表面的吸附构型比(COatop/CObridge)与N-arylpyridinium衍生的二聚体中氮原子的供电子能力(Bader charge)的关系
同时,作者通过DFT计算发现N-arylpyridinium衍生物增强CO的吸附,并且对*COatop的增强程度比对*CObridge的增强更大,从而提高了两种吸附构型比(COatop/CObridge)。N-arylpyridinium衍生物将电子密度转移至*CO周围的水分层,从而更利于增强COatop吸附。同时,Cu(111)表面的DFT计算表明*COatop和*CObridge(C-C)偶联形成二聚物(CO2还原至乙烯的中间产物)是能量最低路径,如图5。但是,太强的CO吸附会反而增加C-C偶联的能垒,从而不利于乙烯的生成。
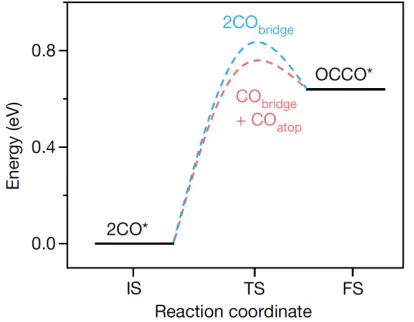
▲图5 *COatop与*COatop偶联时的能垒和*COatop与*COabdrige偶联时的能垒对比
得出结论(理论)
至此,作者用实验和理论计算确定了N-arylpyridinium衍生物影响CO2电还原的关键因素及影响机制,即其给电子能力增强CO吸附强度并调节CO吸附构型比,从而影响*CO偶联的能垒,进而影响乙烯的选择性。
验证理论——基于推导出的理论开发更高效的材料
由于N-arylpyridinium衍生物的给电子能力主要来自N原子,因此,作者推测增加N-arylpyridinium衍生物中的N原子位点并优化其给电子能力,理论上即可得到更高的乙烯选择性。基于该设想,作者合成了一种具有两个N原子位点的N-arylpyridinium,并且其衍生物的给电子能力和对应的CO吸附构型比(COatop/CObridge)都与之前最优N-arylpyridinium的接近,同时,其乙烯选择性也处于火山模型峰顶。因此,作者基于上述理论开发了一个性能更优的N-arylpyridinium盐添加剂,验证了上述理论。
尚未回答的问题
1. 该工作只证明了N-arylpyridinium衍生物可以调节CO的吸附,从而增强C-C偶联,但这很难直接推出其一定提高乙烯的选择性,因为C-C偶联二聚体是CO2还原至乙烯和乙醇共同的中间物,正文中未提及乙醇选择性变化,从supplementary information中的数据可看出乙醇及其他C2+产物的选择性几乎没变化。之前的工作中[1],N-arylpyridinium衍生物将乙醇选择性从7%提至30%,同时将乙烯选择性从14%提至40%。因此,N-arylpyridinium衍生物对乙醇选择性的影响仍是一个开放的问题。
2. 文中未说明为什么DFT计算采用Cu(111)晶面,因为文中的DFT计算基于前一篇工作中提出的反应机理,而那个工作中使用的是Cu(100)晶面 [3],并且Cu(100)也最常用于CO2电还原计算。同时,多晶铜在CO2电还原条件下会重构为Cu(100)晶面 [4]。
参考文献
[1] Z. Han, R. Kortlever, H.-Y. Chen, J.C. Peters, T. Agapie, CO2 Reduction Selective for C≥2 Products on Polycrystalline Copper with N-Substituted Pyridinium Additives, ACS Central Science, 3 (2017) 853-859.
[2] F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C.M. Gabardo, A. Ozden, C.T. Dinh, J. Li, Y. Wang, J.P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C.P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y.C. Li, Z. Han, R.D. Britt, D. Sinton, T. Agapie, J.C. Peters, E.H. Sargent, Molecular tuning of CO2-to-ethylene conversion, Nature, (2019)。
[3] Y. Lum, T. Cheng, W.A. Goddard, J.W. Ager, Electrochemical CO Reduction Builds Solvent Water into Oxygenate Products, Journal of the American Chemical Society, 140 (2018) 9337-9340.
[4] Y.-G. Kim, A. Javier, J.H. Baricuatro, D. Torelli, K.D. Cummins, C.F. Tsang, J.C. Hemminger, M.P. Soriaga, Surface reconstruction of pure-Cu single-crystal electrodes under CO-reduction potentials in alkaline solutions: A study by seriatim ECSTM-DEMS, Journal of Electroanalytical Chemistry, 780 (2016) 290-295.